 Overview of tradeSeq functionality.
Overview of tradeSeq functionality.Abstract
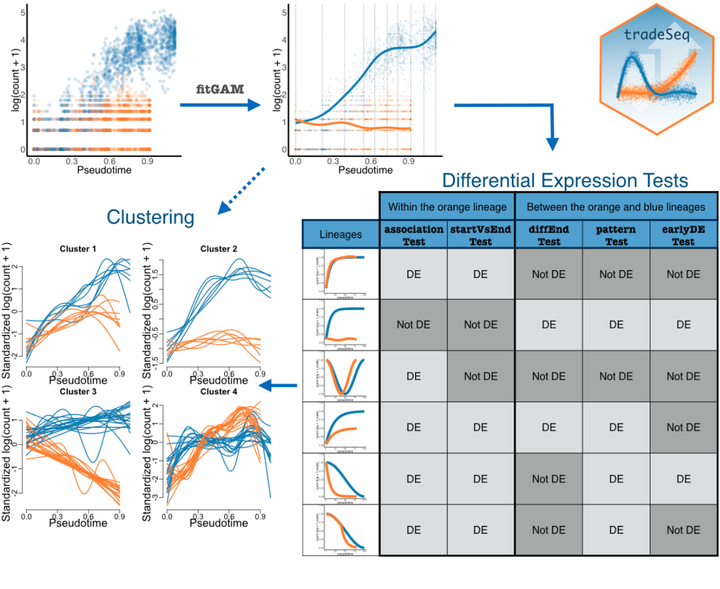
Trajectory inference has radically enhanced single-cell RNA-seq research by enabling the study of dynamic changes in gene expression levels during biological processes such as the cell cycle, cell type differentiation, and cellular activation. Downstream of trajectory inference, it is vital to discover genes that are associated with the lineages in the trajectory to illuminate the underlying biological processes. Furthermore, genes that are differentially expressed between developmental/activational lineages might be highly relevant to further unravel the system under study. Current data analysis procedures, however, typically cluster cells and assess differential expression between the clusters, which fails to exploit the continuous resolution provided by trajectory inference to its full potential. The few available non-cluster-based methods only assess broad differences in gene expression between lineages, hence failing to pinpoint the exact types of divergence. We introduce a powerful generalized additive model framework based on the negative binomial distribution that allows flexible inference of (i) within-lineage differential expression by detecting associations between gene expression and pseudotime over an entire lineage or by comparing gene expression between points/regions within the lineage and (ii) between-lineage differential expression by comparing gene expression between lineages over the entire lineages or at specific points/regions. By incorporating observation-level weights, the model additionally allows to account for zero inflation, commonly observed in single-cell RNA-seq data from full-length protocols. We evaluate the method on simulated and real datasets from droplet-based and full-length protocols, and show that the flexible inference framework is capable of yielding biological insights through a clear interpretation of the data.
Robrecht Cannoodt
Data Science Engineer
R is a horrible language, and I love it (ever since tidyverse, at least).